2018.02.26 Mon |
DeepLearningによる化合物生成(薬、有機発光分子)

最近、DeepLearningを用いた、化合物の合成の論文をよく見かけるので、そのうちの特に有名な論文をご紹介します。
この記事の対象読者層としては、DeepLearning×医学・生物学、DeepLearning×化学、DeepLearning×Material、に興味がある方。
より具体的に言いますと、DeepLearningを用いた化合物(化学構造式)の生成に興味がある方、特定の疾患に対する分子のスクリーニングをしたい方、など、となります。
今回ご紹介する論文は、2016年10月に、Harvard大学、Toronto大学、Cambrige大学、Twitter社の研修者たちによって提出された、
Automatic chemical design using a data-driven continuous representation of molecules
https://arxiv.org/pdf/1610.02415v1.pdf
という論文です。
この論文は、近年のAutoencoderを用いた、分子式の生成モデルの先駆けとなる論文です。
それでは論文紹介を始めていきます。
【論文要約】
分子の離散表現を多次元連続表現に変換する方法と、多次元連続表現から分子の離散表現に変換する方法を報告する。この生成モデルは、化学化合物の自由空間を通じた効率的な検索および最適化を可能にする。
我々は、エンコーダとデコーダを組み合わせて、数十万もの既存の化学構造のdeep neural network用いて訓練した。エンコーダは、分子の離散表現を実数値連続ベクトルに変換し、デコーダは、これらの連続ベクトルをこの潜在空間から離散表現に変換し直した。
連続表現は、ランダムベクトルの解読、既知の化学構造の撹乱、分子間の補間など、潜在空間で簡単な操作を実行することによって、新規な化学構造を自動的に生成することを可能にする。
連続表現はまた、強力な、勾配に基づいた最適化を使用して、最適化された機能性化合物の探索を効率的に誘導することを可能にする。薬物様分子および有機発光ダイオードの設計における我々の方法を示す。
【Introduction(導入・背景)】
薬物および材料設計の目標は、様々な測定可能な目標を最適に達成する新規分子を提案することである。 しかしながら、分子空間における最適化は、探索空間が大きく、離散的で、構造化されていないため、非常に困難である。 新しい化合物を作って試験するには、コストがかかり時間がかかるし、潜在的な化合物候補の数はものすごく多い。 潜在的な薬物様分子の一般的に報告されている範囲は10の23乗~10の60乗であるのに対し、約10の8乗個の物質しか合成されていない。
コンピュータによる計算はこの検索をスピードアップする。数千〜数百万の候補を含むライブラリはコンピュータ計算によりアッセイされ、最も有望な化合物が選択され、実験的にテストされる。
しかしながら、正確なシミュレーションを用いても、計算分子設計は化学空間を探索するために利用可能な探索戦略には限界がある。 現在の方法は、固定ライブラリを介した徹底的な検索か、遺伝的アルゴリズムまたは同様の離散的補間技術のような離散的局所探索方法である。
これらの技術は有用な新規分子をもたらしたが、両方のアプローチは依然として大きな課題に直面している。 固定ライブラリは巨大であり、完全に探索するには多額の費用を必要とし、非現実的な化学反応を避けるために手作業による制御が必要である。 化合物の遺伝的生成は、突然変異および交叉規則のために、発見を助ける経験的で手動の仕様を必要とする。離散最適化法は、勾配などの幾何学的手掛かりを利用できないので、化学空間の広い領域を効果的に探索することが困難である。
分子グラフの表現は、この課題の中心にある。 分子表現は離散的である。それらは、ノードを原子とし、エッジを結合とする無向グラフか、または原子の三次元配列とする無向グラフのいずれかとみなすことができる。 化学情報学の(cheminformatic)目的のために、これらの表現は、理想的には、回転的および並進的に不変でなければならない数値表現に通常変換される。 グラフから得られた表現には、化学指紋、グラフ上の畳み込みネットワーク、類似のグラフ畳み込み、または bag-of-bondsアプローチが含まれる。 クーロン行列アプローチ、散乱変換、原子間距離。 。 。 手頃な価格の理論で得られた3D幾何に基づいている。 ニューラルネットワークに基づく最近開発された表現でさえ、最適ベクトルから分子を再現することはできない。
微分可能、可逆的、およびデータ駆動式の表現は、既存のシステムよりもいくつかの利点がある。第一に、人手を必要とする突然変異の規則は不要であり、ベクトル表現とデコーディングを変更することによって新しい化合物を自動的に生成することができる。大規模な化学データベースには、通常、数百万の分子が含まれているが、ほとんどの特性はほとんどの分子に関して未知である。データ駆動型表現は、大量の非標識化学化合物を活用して、より大きく暗に示されたライブラリーを自動的に構築し、より小さなラベル付きサンプルセットを使用して、連続表現から所望の特性への回帰モデルを構築することができる。微分可能な表現を用いることにより、幾何学的情報を活用して化学空間においてより大きなジャンプを行うための勾配ベースの最適化の使用が可能になる。ベイジアン最適化手法を使用して、
大域的最適性について情報を与える可能性が高い化合物。これらのメソッドは、新しい化合物を提案し、そのプロパティをテストし、この新しい情報を使用してさらに優れた化合物を提案する閉ループに組み込むことができる。
最近の機械学習の進歩により、実際の事例を訓練した後、現実的な合成サンプルを生成できる、強力な確率論的生成モデルが得られた。このようなモデルは、通常、モデル化されるデータの低次元連続表現を生成し、自然画像、テキスト、スピーチ、および音楽に対する補間または類似推論を可能にする。
この研究では、データ駆動のベクトル値表現の分子を構築することによって、新規化合物を生成する連続最適化の使用を提案する。我々は、オートエンコーダーとして訓練された一対のニューラルネットワークを使用して離散表現と連続表現の間で変換を行った。 薬物様分子および有機発光ダイオード(OLED)の設計にこの技術を適用した。
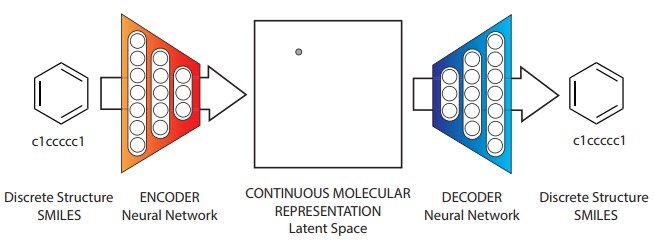
図1: 分子設計のための提案されたオートエンコーダーの図。 SMILESストリングのような離散的分子表現から出発して、エンコーダーネットワークは各分子を潜在空間のベクトルに変換する。このベクトルは効果的な連続分子表現である。 潜在空間内の点が与えられると、デコーダネットワークは対応するSMILESストリングを生成する。https://arxiv.org/pdf/1610.02415v1.pdf
【Methods(方法)】
・分子の初期表示
連続表現にするためのエンコーダーを構築する前に、エンコーディングの前と後の両方で分子を表現するために使用する離散表現を選択する必要がある。テキストをモデリングするためのSeq2Seqオートエンコーダーの最近の進歩の力を利用するために、我々は有機分子によく使われるテキストエンコーディングであるSMILES表現を使用した。 InChIも代わりの文字列表現として試したが、SMILESよりも著しく悪いことがわかった。おそらく、計算と算術を含むより複雑な構文が原因であろう。
・オートエンコーダーの学習
文字列ベースの分子表現の大規模なライブラリから出発して、各文字列を固定次元ベクトルに変換するエンコーダネットワークと、ベクトルを文字列に変換するデコーダネットワークのペアをcomplementaryリカレントニューラルネットワークのペアとして訓練した。このようなエンコーダ・デコーダ対は、オートエンコーダとして知られている。オートエンコーダは、元のストリングを再生する際のエラーを最小限にするように訓練される。すなわち、それはアイデンティティ関数を学習しようとする。オートエンコーダーの重要な洞察は、このアイデンティティマップを情報のボトルネックにさらすことである。このボトルネック(ここでは固定長の連続ベクトル)は、データ内で統計的に最も顕著な情報を捉え、圧縮された表現をネットワークが学習するよう誘導する。 我々はベクターでコードされた分子を分子の潜在的な表現と呼んでいる。SMILES表現の文字ごとの性質と、その内部構文(開閉サイクルと分岐、許容原子価など)の相対的な脆弱性によって、デコーダからの無効な分子が出力されることがある。 私たちは、オープンソースのケミインフォマティクスの一式であるRDKitを使用して、化学産出分子を検証し、無効な分子を廃棄する。
分子設計を可能にするために、自動エンコーダーの連続表現でコード化された化学構造は、最適化する必要がある標的特性と相関させる必要がある。したがって、オートエンコーダーの結果に基づいて、分子の潜在的表現に基づいて分子の性質を予測する第3のモデルを訓練する。有望な新しい候補分子を提案するために、符号化(エンコード)された分子の潜在的なベクトルは、望まれた属性を改善する可能性が最も高い方向に移動され、これらの新しい候補ベクトルが復号化(デコード)される。

図2:中央の分子から出発して、潜在的な空間内で2つのランダムな単位長ベクトルを追跡し、ますます大きな変位を追跡した。 これは、56次元の潜在空間におけるランダムな2次元平面を定義する。 この2次元部分空間内の各位置において、潜在空間内のその点で最も解読される可能性のある分子を示す。 近くの点は類似の分子にデコードされ、遠方の点はさまざまな化合物にデコードされる。
https://arxiv.org/pdf/1610.02415v1.pdf
・オートエンコーダーの構造
文字列は、リカレントニューラルネットワーク(RNN)を使用してベクトルにエンコードできる。 エンコーダRNNをデコーダRNNとペアにして、Seq2Seq学習を実行することができる。また、文字列エンコーディングに畳み込みネットワークを使用して実験を行い、パフォーマンスの向上を確認した。 これは、化学的部分構造、例えば環および官能基に対応する、反復的に並進的に不変の部分文字列の存在によって説明される。SMILESテキストエンコーディングは空白(短い文字列は、残りのところを空白で埋める)を含む35の異なる文字を使用する。最大120文字の文字列をエンコードした。VAEディープネットワークの構造は以下の通りである。エンコーダは、フィルタサイズ9,9,11および畳み込みカーネルはそれぞれ9,9,10の3つの1D畳み込みレイヤを使用した。2つの全結合層(435次元と292次元)が続いている。デコーダは、292次元の全結合層から始まり、隠れ層である501次元の3層の gated recurrent unit networksに供給される。我々はKerasとTheanoパッケージを使用して、 このモデルを訓練した。
RNNデコーダの最後の層は、SMILES文字列内の各位置でのすべてのありうる文字に対する確率分布を定義する。これは、書き込み操作が確率的であり、文字をサンプリングするために使用されたランダムなシードに応じて、同じ潜在ポイントが異なるSMILES文字列にマップされる可能性があることを意味する。
・分子のベイジアン最適化
分子の特徴ベクトルから各分子のコストを予測するために、誘発点500個で疎ガウスプロセス(GP)モデルを訓練した。その後、期待される改善(EI) 発見的教授法を使用して、ベイジアン最適化の10回の反復を実行した。各反復において、EI獲得関数を順次最大化することによって、50個の潜在特徴ベクトル(この塊をバッチと呼ぶ)を選択する。バッチ選択プロセスで未決定の評価を考慮するには、Kriging Believer Algorithmを使用する。つまり、バッチ内の新しいデータポイントをそれぞれ選択した後、そのデータ点をGP予測分布の平均に等しい目標変数に関連する疎なGPモデルの新しい誘導点として追加する。50個の潜在特徴ベクトルの新しいバッチが選択されると、バッチ内の各ポイントは、デコーダネットワークを使用して対応するSMILESストリングに変換される。 SMILES文字列から、(1)を使用して対応するスコア値を取得する。
【Results(結果)】
VAE(variational autoencoders)を使用してコンパクトな表現を作成した。
潜在空間で無制約最適化を実行するには、潜在空間内のほとんどの点が有効なSMILES文字列にデコードされる必要がある。 しかし、自動エンコーダーのトレーニング目的は、潜在的に無効または無意味なSMILESストリングにデコードする潜在的なスペースに大きな「デッドエリア」をもたらすこの制約を強制しない。
潜在空間内のすべての点が有効な分子に対応することを確実にするために、我々は
私たちのオートエンコーダーとその目的をVAEに変更した。 VAEは、各データポイントが対応するが未知の潜在的表現を有する潜在変数モデルの原理的近似推論方法として開発された。 VAEはオートエンコーダを一般化し、エンコーダに確率性を追加し、潜在空間のすべての領域を有効なデコードに対応させるペナルティ項を追加する。直感的には、符号化された分子にノイズを加えると、デコーダはより多様な潜在ポイントをどのように復号するかを覚えるようになる。さらに、2つの異なる分子は確率論的に潜在空間に閉じ込められた符号化を有することができるが、依然として異なる分子グラフに解読する必要があるので、重複を避けるために符号化が潜在空間全体に広がることも奨励する。 RNNエンコーダおよびデコーダネットワークを有するVAEを使用することは、Bowmanらのアプローチに近づいている。オートエンコーダーは、ZINCデータベースから抽出された約250,000の薬物様市販分子でデータセットを訓練した。我々はまた、計算的にのみ生成された約100,000個のOLED分子についてこのアプローチを試験した。
最適なディープオートエンコーダ構成を検索するために、複数のハイパーパラメータ(RNNまたはCNNエンコーダ、隠れ層の数、レイヤサイズ、正規化、学習率など)に対するベイズ最適化を実行した。 また、最適な外部ループを実行して、潜在的な次元がどれほど小さくても、妥当な再構成エラーを生成するかを決定した。 薬剤感受性分子に関して、ナイーブでVariationalなオートエンコーダーで訓練した後、同じ構造を有機発光(OLED)分子に対しても使用した。
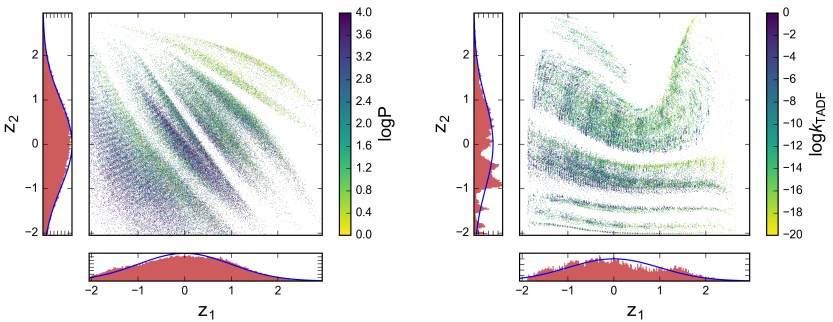 図3:学習された2次元潜在空間への分子訓練セットの投影。 1次元ヒストグラムは、各次元に沿ったトレーニングデータの分布を示し、事前にVAEに課されたガウス分布でオーバーレイされる。 これらの点は、それらの機能に関連する化学的性質に沿って着色され、最適化実験の標的となる。 左:薬剤と同様の分子の天然ライブラリーで、予測される水 – オクタノール分配係数によって着色されている。 右:有機LED分子の誘導体群的に生成されたライブラリーで、予測された遅れた蛍光放出速度(μT-1でkTADF)で着色されている。
図3:学習された2次元潜在空間への分子訓練セットの投影。 1次元ヒストグラムは、各次元に沿ったトレーニングデータの分布を示し、事前にVAEに課されたガウス分布でオーバーレイされる。 これらの点は、それらの機能に関連する化学的性質に沿って着色され、最適化実験の標的となる。 左:薬剤と同様の分子の天然ライブラリーで、予測される水 – オクタノール分配係数によって着色されている。 右:有機LED分子の誘導体群的に生成されたライブラリーで、予測された遅れた蛍光放出速度(μT-1でkTADF)で着色されている。
図3は、次元の潜在空間を持つオートエンコーダーを訓練した結果を示している。 薬物様分子の場合、それらは多様で自然な訓練セットであるため、分子が空間全体に均一に広がっており、それらは両方の次元に沿って非常に密接にガウス型に従うことが観察された。 一方、OLED分子は、たとえ2次元であっても、密接にクラスター化され、潜在的な表現においてより大きなギャップを残した。 符号化された分子の分布は、効果的に正規化されてガウス形状にならず、各次元に沿って高度に構造化されていた。
これは、それらが生成されたコンビナトリアル・ドナー・ブリッジ・アクセプタ方式(combinatorial donor-bridge-acceptor way)の結果であり、オートエンコーダが潜在的な表現を効率的に学習することの難しさを説明している。
両方の予測において、正則化された薬物様セットでさえ、我々は、複数のスケールでの2D空間におけるbanding(バンド形成?)および構造を観察する。
これは、ディープ・オートエンコーダーが分子の類似性に対処し、離散的なものから連続的なものへとマッピングする能力を示している。興味深いことに、プロットされた分子特性は、潜在的な座標に著しい依存性を示す。エミッター(emitter)ライブラリーの場合、局所的およびより一般的な規模の両方で、類似した特性を示す傾向がある。
類似の分子が類似の性質を有することは、化学における構造 – 特性関係の基礎であり、任意の分子表現のための望ましい特徴である。オートエンコーダは、ターゲットプロパティとは無関係に監督されていない方法で訓練されているため、この結果は特に有益である。
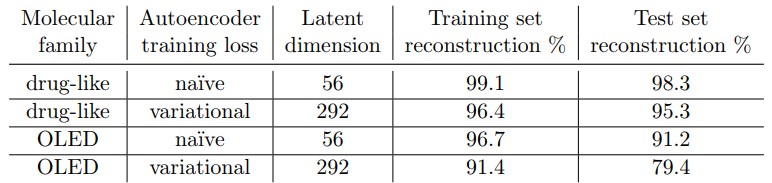 表1:本研究で使用したディープオートエンコーダの再現精度。 精度は、デコードされたSMILES文字列の正しい文字のパーセンテージとして定義されている。 十分な潜在次元を持つオートエンコーダーは完全な再構成を達成できるが、潜在次元の増加に伴い潜在空間の探索がより困難になる傾向がある。
表1:本研究で使用したディープオートエンコーダの再現精度。 精度は、デコードされたSMILES文字列の正しい文字のパーセンテージとして定義されている。 十分な潜在次元を持つオートエンコーダーは完全な再構成を達成できるが、潜在次元の増加に伴い潜在空間の探索がより困難になる傾向がある。
表1は、私たちの最善の薬とOLEDオートエンコーダーが、ナイーブとバリエーションの両方の構成で学習セットとテストセットを再構築する能力を比較し、機械生成OLEDライブラリで潜在的な表現を学ぶ大きな課題を確認する。
分子空間における摂動と補間
我々は、分子構造の符号化と復号化を含む多くのタスクにおいて、VAEの性能を分析した。
図10と図11は、潜在空間内のランダムにサンプリングされた8つのポイントの複数のデコードを示している。
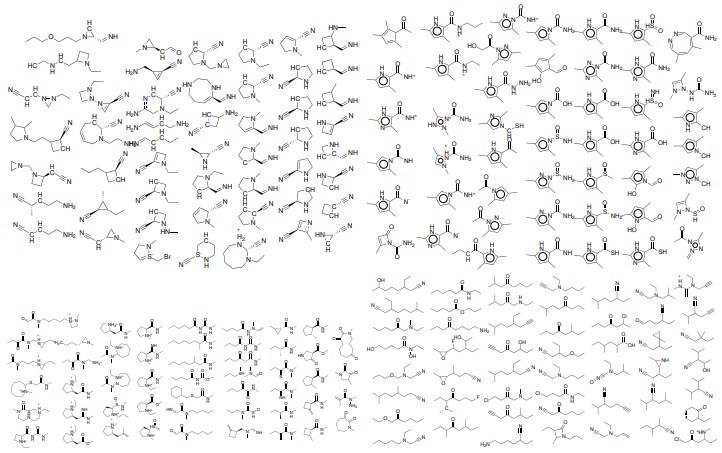 図10:VAEの潜在空間内のランダムサンプリングされた点からデコードされた分子。
図10:VAEの潜在空間内のランダムサンプリングされた点からデコードされた分子。
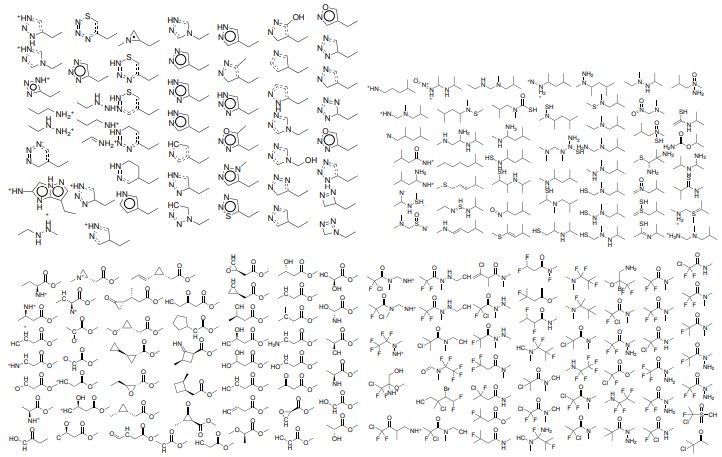 図11:VAEの潜在空間における無作為抽出点からデコードされた分子。
図11:VAEの潜在空間における無作為抽出点からデコードされた分子。
ランダムな点は中規模の現実的な分子にデコードされ、潜在空間の点が有効な分子にマップされることを保証する上でVAEが有効であることを示唆している。 潜在空間における各点についてデコードされた分子内のわずかな変化は、デコーダの確率論的性質によるものである。
1386個のFDA承認医薬品からのランダムに薬を選択し、VAEをサンプリングしてコード化し、それらの潜在的発現を解読した。 図4、図8および図9はそのようなプロセスの結果を示す。
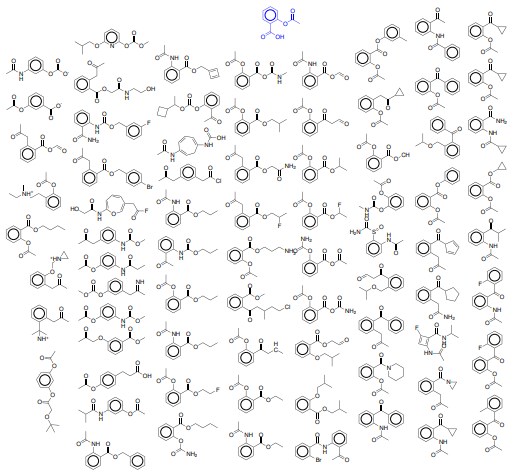 図4:特定の分子(アスピリン[2-(アセチルオキシ)安息香酸](aspirin [2-(acetyloxy)benzoic acid])の近く、変色オートエンコーダの潜在空間のランダムにサンプリングされた点から解読された分子、青色で強調表示)。
図4:特定の分子(アスピリン[2-(アセチルオキシ)安息香酸](aspirin [2-(acetyloxy)benzoic acid])の近く、変色オートエンコーダの潜在空間のランダムにサンプリングされた点から解読された分子、青色で強調表示)。
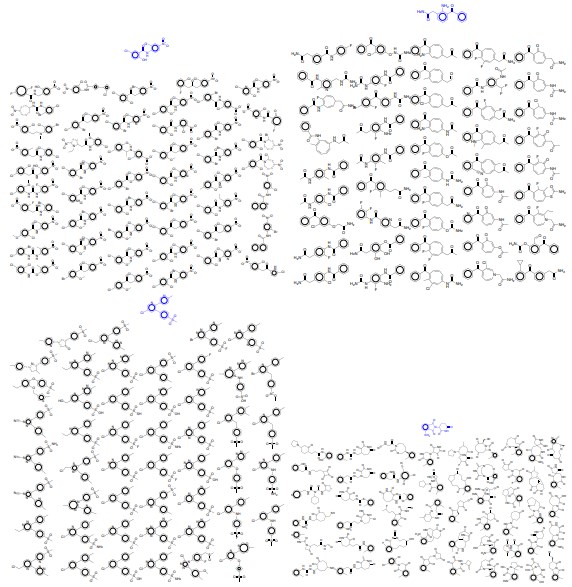 図8:コード化分布からの複数のサンプルのデコードを伴って提示された、VAEへの入力として使用された分子。
図8:コード化分布からの複数のサンプルのデコードを伴って提示された、VAEへの入力として使用された分子。
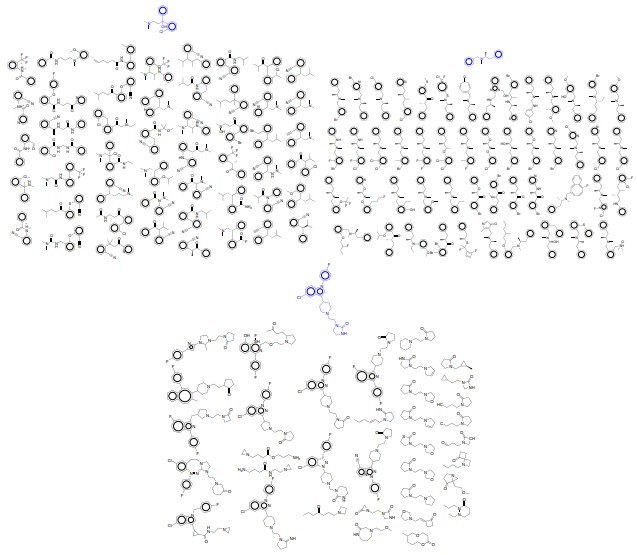 図9:変分的オートエンコーダーへの入力として使用された青色の分子で、コード化分布からの複数のサンプルの解読が提示された。
図9:変分的オートエンコーダーへの入力として使用された青色の分子で、コード化分布からの複数のサンプルの解読が提示された。
VAEの確率論的サンプリングとSMILESストリング解読の確率的性質の両方の結果として、元の化合物の複数の化学的変化が得られた。
潜在空間におけるサンプリング点に加えて、潜在空間におけるランダムな方向の意味を分析した。 ランダム薬物分子から出発して、潜在空間における2つのランダムな単位ベクトルを追跡し、分子にデコードした。 図2-13は、中心の開始分子と、水平および垂直方向の外挿分子の最も可能性のあるデコードを示している。 潜在空間のほとんどの点は、現実的な薬物様分子に対応する。 関連する実験では、画像の他の生成モデルの成功に続いて、我々は化学空間で補間を行った。 FDAで承認された分子のリストからランダムな薬物が選択され、VAEの平均をサンプリングすることによってコード化された。 その後、2次元で線形補間を行った。 潜在空間内の各点を複数回デコードし、潜在的表現が一度再エンコードされると、サンプリングされた点に最も近いものを報告した。 (図14-15)
図14:補間。 4つのランダム薬間の2次元補間。 左:潜在空間内の場所の四隅をコード化した開始分子。
右:4つの分子の潜在的表現間の線形補間点の解読。
 図15:補間。 4つのランダム薬間の2次元補間。 左:潜在空間内の場所の四隅をコード化した開始分子。
図15:補間。 4つのランダム薬間の2次元補間。 左:潜在空間内の場所の四隅をコード化した開始分子。
右:4つの分子の潜在的表現間の線形補間点のデコード。
薬物様分子のベイジアン最適化
提案された分子オートエンコーダーを使用して、所望の特性を有する新しい分子を発見することができる。
簡単な例として、RDkitによって推定されるように、最初に水 – オクタノール分配係数(logP)を最大にすることを試みる。 logPは、分子のドラッグライクネスを特徴づける重要な要素であり、薬物設計に目的がある。 得られた分子が実際に合成しやすいことを確実にするために、合成アクセシビリティ(SA)スコアも我々の目的に組み入れる。
logPとSAスコアのみを最適化した当初の実験では新規分子が生成されたが、非現実的に大きな炭素原子の環を持つ分子が生成された。 この問題を避けるために、私たちは、私たちの目標に6以上のサイズのカーボンリングを持つことに対するペナルティを加えた。
目的変数J(m)は、分子mに対して、
J(m) = logP(m) − SA(m) − ring-penalty(m) ・・・(1)
と定義できる。
スコアlogP(m), SA(m),およびリングペナルティ(m)は、訓練データにわたってゼロ平均および単位標準偏差を有するように正規化される。
上記プロセスによって選択された500個の潜在特徴ベクトルの半分以上が有効なSMILES文字列を生成した。 得られた分子の中で、2つの最良値は、5.02および4.68の目的変数の値を有し、訓練データにおける最も高い目標変数の値、4.52より高かった。 図7の右部分は、トレーニングデータ中の分子の目的変数の値の経験的分布を示す。 2つの新しい分子は、図7の左側に示されている。
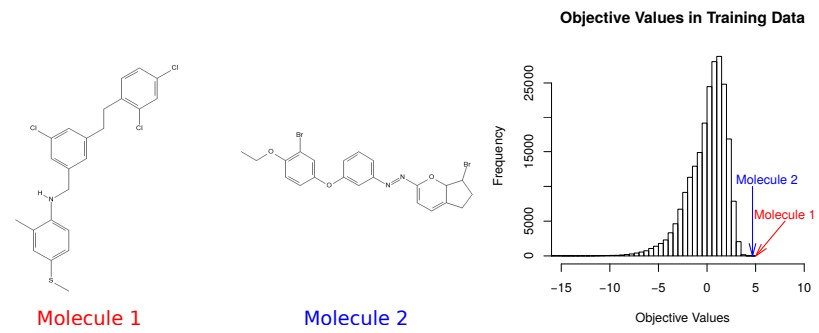 図7:左:最適化プロセスによって生成された分子で、訓練データ中の他のどの分子よりもスコア値が高い。 右:訓練データの目的変数の値のヒストグラム。
図7:左:最適化プロセスによって生成された分子で、訓練データ中の他のどの分子よりもスコア値が高い。 右:訓練データの目的変数の値のヒストグラム。
目的変数(1)の高い値は必ずしも高いlogPスコアに変換されるとは限らない。
しかし、図7の分子のlogPスコアは8.07と8.51であり、トレーニングデータの最高logPスコアは8.25である。 したがって、第2の分子は、トレーニングセット中の他の分子よりも高いlogPスコアを有する。 これは、分子オートエンコーダーをベイジアン最適化と組み合わせて、トレーニングセットにあるものよりも優れた特性を持つ新しい分子を発見できることを示している。
有機発光物質の実験
有機エレクトロニクスの分野は固体状態無機材料の代替物としての有機分子の大きな設計可能性と有望な性質のために、特に有機光におけるシミュレーションベースの分子設計において非常に活発である。大きな設計可能性と有望な特性有機エレクトロニクスの分野は、特に有機発光ダイオードにおけるシミュレーションベースの分子設計において非常に有効である。 OLEDは、電流に応答して発光することができる有機分子である。それらは現在、小型ディスプレイ、主にスマートフォンに適用されており、液晶ディスプレイを大面積ディスプレイのLEDバックパネルに置き換える新しいディスプレイのパラダイムになる可能性がある。最新のOLED技術である熱アシスト遅延蛍光(TADF)は、エミッタの蛍光特性を、その最低三重項と一重項励起状態との間の低いエネルギー差と組み合わせることに依存する。これらの特性の設計プロキシは、発光色などの特性とともに、単純な量子計算に適しており、最近の様々な研究によって、TADFおよび燐光の両方での新しいOLEDのコンピュータ駆動設計に取り組んでいる。この理由から、このクラスの分子でオートエンコーダーシステムの性能をテストした。
我々は、フラグメントの組み合わせによって生成された約150,000の初期分子を有するトレーニングセットを使用した。時間依存密度汎関数理論を用いて、これらの分子の異なる特性(発光色、遅延蛍光減衰速度(kTADF)など)を推定した。その後、我々は潜在空間でこれらの特性を最適化することによって新しい分子を作り出した。残念なことに、このケースでは、ベイジアン最適化手順によって選択された潜在的なベクトルが有効なSMILE文字列を生成しなかったか、または結果のSMILESが既にトレーニングデータに見つかった。この場合、分子オートエンコーダーは、化学空間の一般的な潜在的表現を学ぶことに失敗したと考えられる。これはおそらく、オートエンコーダーを訓練するために使用された分子がお互いに非常に類似しているという事実によると思われる。これは、表1に報告されている精度の低下をもたらした。これらの問題は、多量の未標識分子を用いて、分子オートエンコーダーを訓練することによって潜在的に対処することができた。
【限界(Limitation)】
現在のtwo-stage learningアプローチ(実際の薬分子や発光分子であるラベル付きデータを学習させた後に実際の薬でない分子や発光しない分子を学習させることか、EncoderとDecoderを使っていることのどちらかの意味と思われる)の1つの問題は、教師なし学習からの潜在的表現が、最適化されている特性に円滑にマッピングされない可能性があることです。 この問題に対処する直接的な方法は、両方の目的を共同して鍛えることである。共同トレーニングはモデルが簡単にデコードされ、予測が容易な潜在的な表現を見つけることを促すだろう。 加えて、より多くのデータを持つより深いオートエンコーダーを訓練することによって、より良い一般化を得ることも期待されている。 1億に近い化合物の化学構造が知られており、既知の化学の単一の統一された埋め込みを訓練するために使用することができる。 このタスクには、複数のGPUを使用するソフトウェアパッケージが適用されている。
この作業では、テキストベースの分子コードを使用したが、グラフベースのオートエンコーダーにはいくつかの利点がある。デコーダが有効なSMILESストリングを生成するように強制すると、学習は不必要に困難になる。なぜなら、デコーダが暗黙的にどのストリングが有効なSMILESであるかを覚えておく必要があるためである。分子動態グラフを直接出力するオートエンコーダは、グラフ同型性の問題や有効な分子グラフに対応しない文字列の問題に直接対処するため、魅力的である。分子グラフを取り込むエンコーダは、ECFPや、神経分子指紋(neural molecular fingerprints)などのECFPの連続的にパラメータ化された変種のような市販の分子指紋法を用いて、率直(straight forward)である。しかし、任意のグラフを出力できるニューラルネットワークを構築することは未解決の問題である。文字列エンコーディング手法のもう1つの問題は、デコーダが無効なSMILES文字列を生成することがあることである。オートエンコーダは入力として有効な分子のみが提示されるので、デコーダは無効な分子についての化学的規則の正確な知識を欠いている。 同様の問題に対して、識別器を訓練するために有効なデータと無効なデータ(失敗した実験)の両方を使用することによって、化学実験の結果を機械学習することが行われている。
【議論(Discussion)】
我々は、分子の連続的なコード化に基づいて化学空間を探索するための新しい方法を提案した。 これらの方法は、化合物のライブラリを手作りで作成する必要性を排除し、化学空間を介して新しいタイプの指示された勾配ベース検索を可能にする。 多様な代表データで訓練すると、原物類似性(fidelity)と最適化能力は優れていますが、機械で生成されたコンビナトリアルライブラリでは原物類似性と最適化能力は低い。
最後に、上で取り上げなかった、図を張り付けておきます。
 図5:補間。 4つのランダム薬間の2次元補間。 左:エンコードされた開始分子。そのデコードは右図の四隅に対応。 右の4つの分子の潜在的な表現間を直線的に補間する結果のデコード。
図5:補間。 4つのランダム薬間の2次元補間。 左:エンコードされた開始分子。そのデコードは右図の四隅に対応。 右の4つの分子の潜在的な表現間を直線的に補間する結果のデコード。
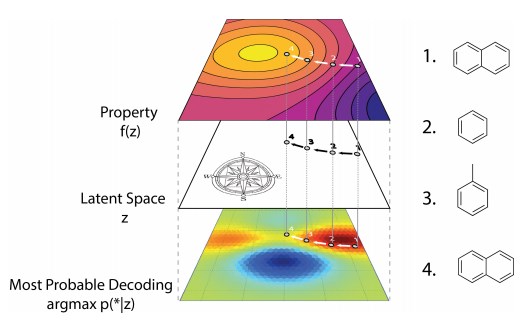
図6:連続潜在空間における勾配に基づく最適化。 それらの潜在的表現zに基づいて分子の特性を予測する代理モデルf(z)を訓練した後、所望の特性の高い値を有すると期待される新しい潜在的表現を見つけるためにzに関してf(z)を最適化することができる。 これらの新しい潜在表現はSMILES文字列にデコードされ、その時点でそれらの特性を経験的にテストすることができる。
ということで、今回、
Automatic chemical design using a data-driven continuous representation of molecules
https://arxiv.org/pdf/1610.02415v1.pdf
のほぼ全体を翻訳・意訳してみました。
この論文は、今後NeuralNetworkを用いて化合物合成を行うなら必読となるような、milestones的な論文です。しかし初心者・異分野の人が理解することは極めて難しいです。なのでとりあえず和訳してみました。和訳した僕自身かなり理解が深まりました。普通に英語のまますべて読んだときが理解度10%なら、全訳したら理解度が60%くらいというくらい飛躍的に理解度が上昇しました。
全部訳すのにおそらく20-30時間はかかっているので(いろいろ調べながらやっているのもありますが)みんながみんなできるとは思いませんが、時間ある人にはお勧めです。
では今回はここまでで。
鈴木瑞人
東京大学大学院新領域創成科学研究科メディカル情報生命専攻 博士課程
実践的機械学習勉強会 代表
株式会社パッパーレ 代表取締役
NPO法人Bizjapan テクノロジー部門BizXチームリーダー