2017.02.25 Sat |
BioinformaticsとDeepLearningの融合分野の論文調査1

今回は、Bioinformaticsの分野において、DeepLearningが使われている例をご紹介したいと思います。
論文のチョイスは独断と偏見に基づいています。
この記事の対象者は、ゲノム情報に対してそれなりの知見をもっていて、Neural Network系の機械学習をそれなりに知っている方となります。知識ゼロの型に方にもできるだけわかりやすくご説明しますが、わかりにくいところもあるかも、しれませんが、ご了承ください。もしも解説内容が間違っていたら、下記目メールアドレスまでご連絡くださると幸いです。
machine.learning.r@gmail.com
それでは早速はじめましょう。
今回ご紹介する論文は以下になります。
 http://genome.cshlp.org/content/26/7/990.long
http://genome.cshlp.org/content/26/7/990.long
Genome Researchといったら、ゲノム界では最も有名なジャーナルの一つです。
では論文の概要からまとめていきましょう。
【概要】
・DeepConvolutionalNeuralNetwork(以下CNN)をもちいて、いろいろやった。
・ゲノムデータからCNNをもちいて、DNA配列の機能予測を可能にするBassetパッケージを作成・公開した(Facebookが作成・公開しているDeepLearningのフレームワークTorch7を使用)。
・DNaseでゲノムを分解してからゲノムを読んで得られるデータ(以下DNase-seq)を用いて、164種類の細胞種の分類を、Bassetをもちいて行い、既存の手法より高い分類能力を示すことに成功した。
・Bassetは、変異配列間の近接可能性の変化の予測に関して、既存のGWASのSNPs(連鎖不平衡の関係にあるSNPs:なんらかの関係があってセットで子孫に受け継がれる確率の高いSNPs)を用いた予測よりもすばらしい結果を出した。
・Bassetを用いて、細胞型の関心領域で単一のシークエンシングアッセイを行い、その細胞のクロマチン近接可能性コードを同時に学び、現在の近接可能性と近接可能性に影響を与えるゲノムのあらゆる突然変異を注釈することができた。
【個人的なコメント】
・CNNをゲノム情報に適用した貴重な先行事例。
・GWASで得られたSNPsを使うのでなく、DNA消化酵素のDNaseをもちいてゲノムを処理してそれをさらに、CNNで教師あり機械学習の分類を行ったところが、二重に新しい。
・後半になるにつれて難しくなっている。
・PhyloPやin silico saturated mutagenesis、PWMという単語が説明なしで使われているあたり、bioinformaticsの論文らしさを感じた。
【導入部分の要約】
・ゲノムと表現型の統計的関係を特定することは、ヒトの健康と疾患にとって非常に重要である。
・非翻訳領域(たんぱく質をコードしていない領域)をわれわれは効率的に解釈できないのは問題である。
・今まで行われていた研究により、教師データがあれば、機械学習の教師あり機械学習により、タンパク質結合、DNAアクセス可能性(ヘテロクロマチンかユークロマチンかどうかなど)、ヒストン修飾、DNAメチル化をその配列から効果的に予測することができるようになった。
・近年、画像解析や自然言語処理において、多層ニューラルネットワークが画期的な進歩を達成している。
・手作業または前処理ステップで特徴量を選択するのではなく、畳み込みニューラルネットワーク(CNN)によって、トレーニング中にデータから適応的に学習する。
・DNA配列解析へのCNNの早期適用は、DNA配列からのタンパク質結合および接近可能性を予測する上で、サポートベクターマシンまたはランダムフォレストなどのより確立されたアルゴリズムを上回る。
・しかしこのCNNの価値を利用するには、このモデルに対して、技術的・概念的に、研究者がアクセス可能でなければならない。
・そこでわれわれは、Bassetパッケージを提供します。Bassetパケージは、DNA配列の機能予測を提供する。
・我々は、例として、ENCODEプロジェクトコンソーシアムおよびロードマップエピゲノムコンソーシアムによってDNase-seqによってマッピングされた164の細胞型におけるDNA配列の接近可能性を同時に予測するためにBassetを使用した。
・ここでは、CNNは、関連する配列モチーフおよび制御ロジックを同時に学習し、細胞特異的DNAアクセシビリティ(どの部位のDNAがほどけているか)を決定した。
・次に、GWASで見つかっている各変異にDNA接近可能性の違いを予測する、細胞種特異的なスコアを与えた。
・Bassetは、ゲノム生物学コミュニティの手にCNNを置き、研究者が新しいデータセットのモデルを訓練し分析するためのツールと戦略を提供する。
【使用データ】
ENCODE Project Consortiumの125種類の細胞型におけるDNase-seqデータ、
Roadmap Epigenomics Consortiumが提供する39種類の細胞型におけるDNase-seqデータ
を使用。
【予備知識】
・DNase I hypersensitive site (DHS):DNAを分解する酵素である、DNaseは、ゲノムのうちほどけている部分(ユークロマチン)に接近し、DNAを分解する。ゲノムのうちほどけている部分は、転写調節領域(エンハンサー、プロモーター、インシュレーター、サイレンサーなど)であることが多く、その細胞における個性のようなもの。DNase-seqでは、前処理として、DNaseを作用させるので、細胞型固有のDHSが分解されたDNA配列が読まれる。
【結果要約】
Fig1では、Bassetで実装されている、Convolutional Neural Networkの構造が示されています。
入力されるゲノム情報は、一塩基(塩基はA,T,C,Gの4種類)が一つのベクトルとして表される(one hot code)。たとえば、Aなら(1,0,0,0)、Tなら(0,1,0,0)、Cなら(0,0,1,0)、Gなら(0,0,0,1)といった具合。 そのベクトルを並べた、one hot code sequenceがゲノム情報となる(ここではゲノム情報とDNA配列はほぼ同じ意味として使う)。
下にFig1として、示されているのは、One Hot Code Sequenceとして、ゲノム情報を、CNNにいれる作業と、CNNの構造を示しています。CNNで何をやっているかまだ未学習の方は、”ゼロから作るDeep Learning”という本をご一読ください。https://www.oreilly.co.jp/books/9784873117584/
CNNの構造は、One Hot Code Sequence→Convolution Layers×3→Fully Conected Layer→Multi-Task Predictionという構造になっている。
入力したゲノム情報は、最終的にMulti-Task Predictionの層で、どの細胞に分類されるかの結果が出るようになっている。

http://genome.cshlp.org/content/26/7/990.long
次の図(Fig2)は、Aで、細胞種ごとのDNase 1 hypersensitive sites(DHS)を表していて(縦軸に細胞種、横軸にDHS)、Bでは既存の機械学習の手法(gkm-SVM)よりも今回のCNNの方が細胞分類の精度が高いことを表していて、Cでは、5種類の細胞種における、ROC曲線とAUCの値(1に近いほど良い予測能)を示している。
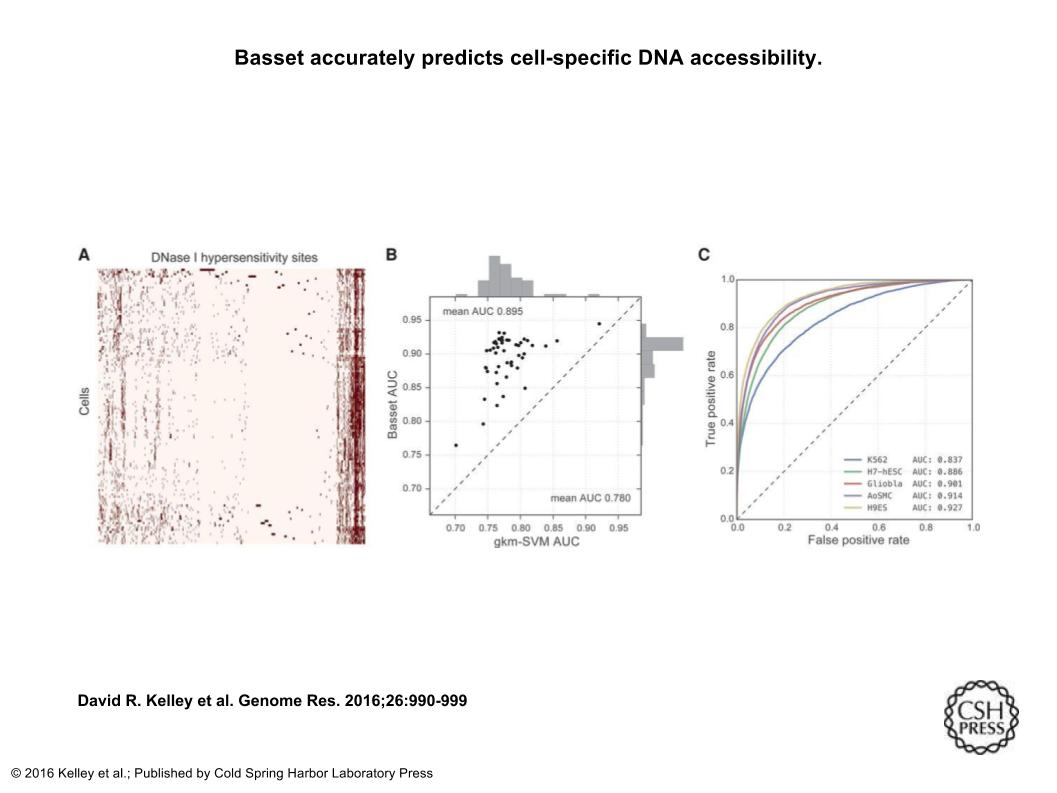
http://genome.cshlp.org/content/26/7/990.long
次のFig3では、Bassetの一番初めの畳み込み層が、既知および新規の配列モチーフを発見したことを表している。Aの散布図では、x軸は、300の第1層畳み込みフィルタによって表されるPWMの情報内容を表し、 y軸は、その平均値にフィルタからのすべての出力を設定し、すべてのセルの上でアクセシビリティ予測変化のベクトルの二乗和を取ることによって計算影響のスコアを算出した値を表している。TomTomモチーフ比較ツールによってヒトCIS-BPデータベースの既知のTFモチーフに0.1のq値で注釈を付けることができるかどうかによって色分けしている。Bでは、45%のフィルタには注釈を付けることができその一部を表している。human CIS-BP databaseのTF(Transcription Factor)motifとBassetが畳み込み層で出力したPWMを表している。Cでは、各細胞種での接近可能性予測の影響度の値で、フィルターをクラスタリングした結果を表しており、下のたくさん並んでいる列名がフィルター名、右横に書いてあるアルファベットが、モチーフ名を表している。このクラスタリングの結果、TP63・GRHL1・KLFといった上皮の発達に関係する因子を抽出するフィルタが特定された。
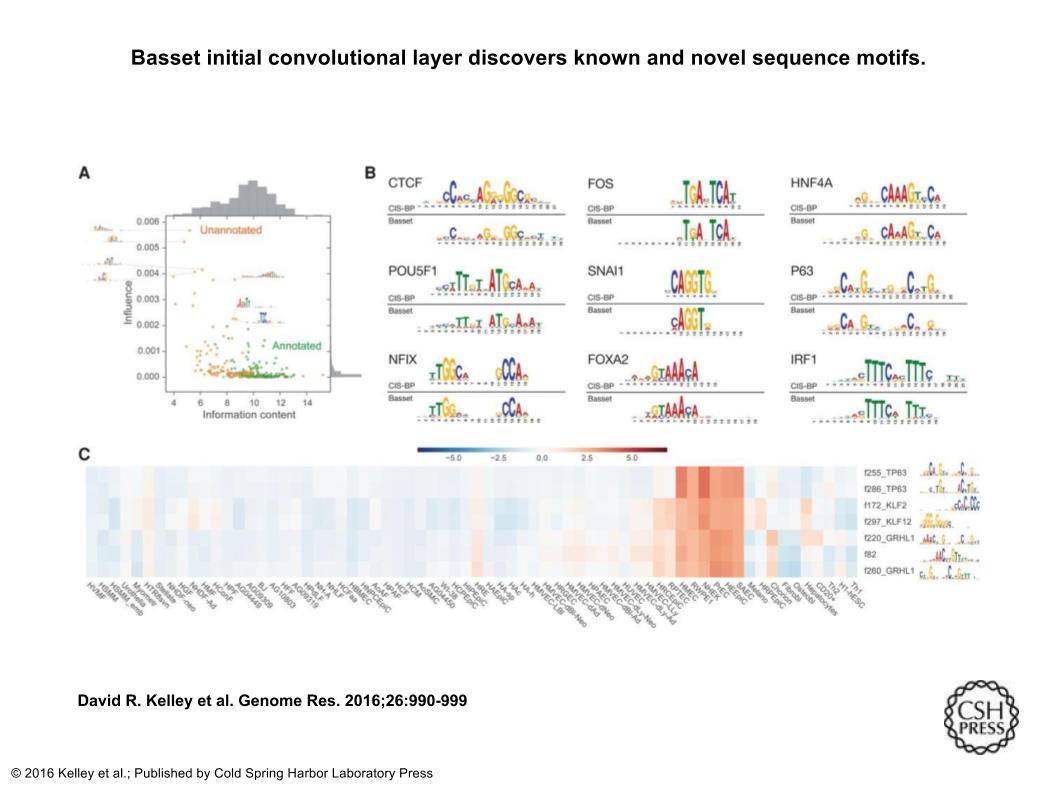 http://genome.cshlp.org/content/26/7/990.long
http://genome.cshlp.org/content/26/7/990.long
次のFig4では、コンピュータ上でのsaturated mutagenesis(アミノ酸が変化するようにヌクレオチドに変異を起こす)の結果を示している。(A)Bassetを用いて細胞株H1-hESCにおけるChr 9:118,434,976-118,435,175領域の接近可能性に対するすべての突然変異の影響を予測した。ヒートマップは、突然変異した配列の予測された接近可能性の変化を示す。各列は、シーケンス内の位置に対応している。各行は、対応するヌクレオチドへの突然変異を表す。損失スコアは、真のヌクレオチドからの全ての変異の中の最大の減少を示し、ゲインスコアは最大増加を示す。ヌクレオチドは最小の高さを超えて、損失スコアに比例するように描かれている。 AP-1複合体を作ることで知られるTGASTCA motif(FOS)の部分が強調されているが、H1-hESCsにおけるJUNとJUNDのChIP-seqがその複合体の結合を確認している。結合したモチーフは、PhyloPに従って高い保存性を示す。損失スコアはPhyloPと強い関係があることもわかる。利得スコアだけではより弱い関係だが、利得スコアと損失スコアの組み合わせが最も強い関係であることがわかる。
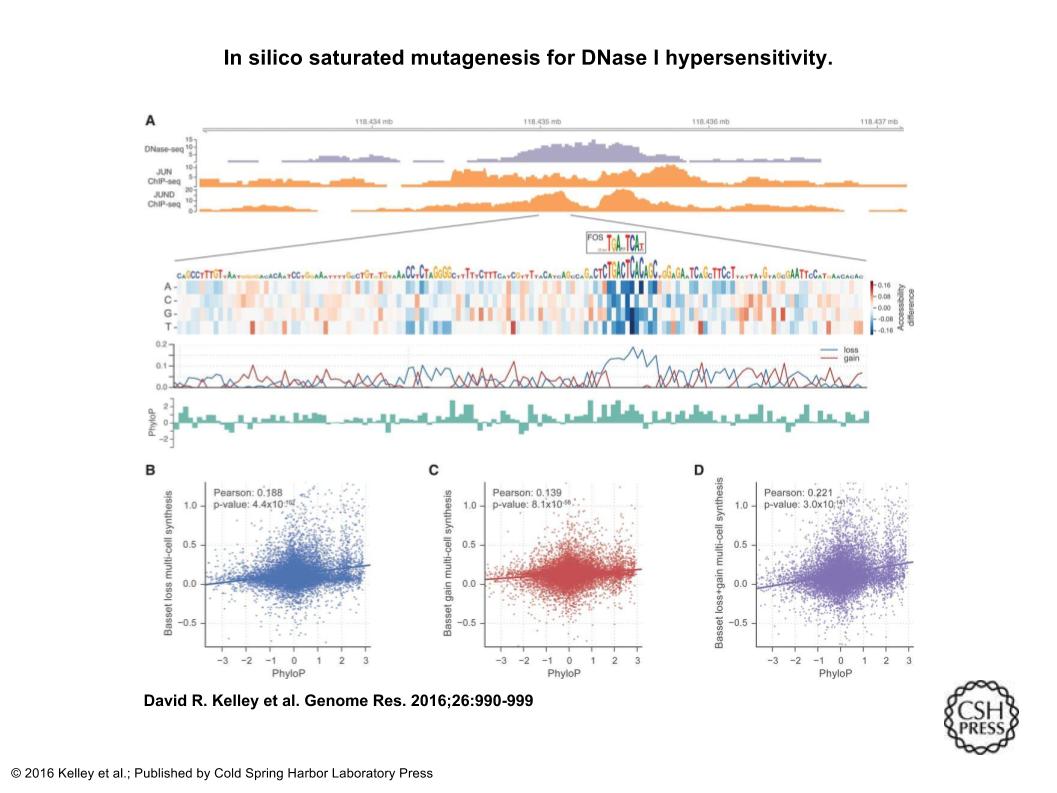
http://genome.cshlp.org/content/26/7/990/F4.expansion.html
自己免疫疾患に関連する8741個のGWAS SNPを、PICSと呼ばれる統計的手法で解析した。PICSとは高密度なgenotypyingデータを用いて、連鎖不平衡(LD)の原因となるSNPsである確率を割り当てるもの。多くの場合、PICSは原因となるSNPを高い確率で特定する。
LDのSNPがタンパク質コード遺伝子に影響を及ぼさない7252個のGWAS SNPに焦点を当て、因果確率が0.5以上の235個の高PICS SNPと、因果確率0.05を割り当てられた3004個の低PICS SNPを分類した。SADプロファイルの平均値は、高PICS SNPのセット(Mann-Whitney U検定、P値<1.3×10 -7)で有意に大きくなった。低PICS SNPよりも7倍以上高いSNPは、細胞型に対して平均して0.1を超える接近可能性を変化させると予測された(Fig5A)。
PICSとの間で、白斑、関節リウマチ、多発性硬化症における免疫機構に関連するrs4409785があった。PICSはrs4409785に、白斑の因果関係の確率を85.3%割り当てている。SNP(rs4409785)は559kbの長さの遺伝子砂漠に位置している。そして、tyrという遺伝子からは、6.28Mb離れている。 しかし、TYRは、皮膚に色を与える色素であるチロシンからメラニンへの変換を触媒するたんぱく質のため、TYRは皮膚色疾患白斑のためのもっともらしいメカニズムを提供すると考えられている。
Bassetの予測はこの仮説を支持している。より一般的なT配列には活性がないが、C配列はモデルのCTCFフィルターによって認識されるモチーフを生成する(Fig5B)。この配列はCTCFデータベースのモチーフと完全に一致しているが、Bassetは、H1-hESCにおいて接近可能性が0.8%から73.24%に増加するなど、すべての細胞タイプで接近可能性が劇的に増加すると予測している。対立遺伝子特異的CTCF結合の実験的証拠を評価するために、我々はENCODE(ENCODE Project Consortium 2012)によって様々な細胞型で実施された88のユニークなCTCF ChIP-seqデータセットをダウンロードし、これらのうち、21の細胞型はrs4409785に整列した3つ以上の読み取りを有し、そのうち11つは有意なピークコールを有していた(Fig5C)
次のFig5は、ゲノム変異の解釈を可能にするSNP Accessibility Difference(SAD)スコアについて書かれている。(A)Bassetを用いて、determined by population fine mapping dataによって決定された近くのSNP(PICS確率<0.05)と比較して、可能性が高い因果GWAS SNP(PICS確率> 0.5)に対して大きなスコアを割り当てた。barは、0.1を超える全ての細胞型にわたってSADプロファイル平均を割り当てられたSNPの割合を測定している。(B)haplotype blockのPICSビューと一致して、最高のSADスコアのうちrs4409785に注釈をつけた。Bassetは、より一般的なT配列が完全に休眠していると予測するが、この領域はBassetがCTCF結合部位のために非常に高い接近可能性を有すると考えられる部位にC配列と変換する。(C)CTCF 88個のユニークな細胞型のChIP-seqは、この部位のCTCFの対立遺伝子特異性を強く支持する。
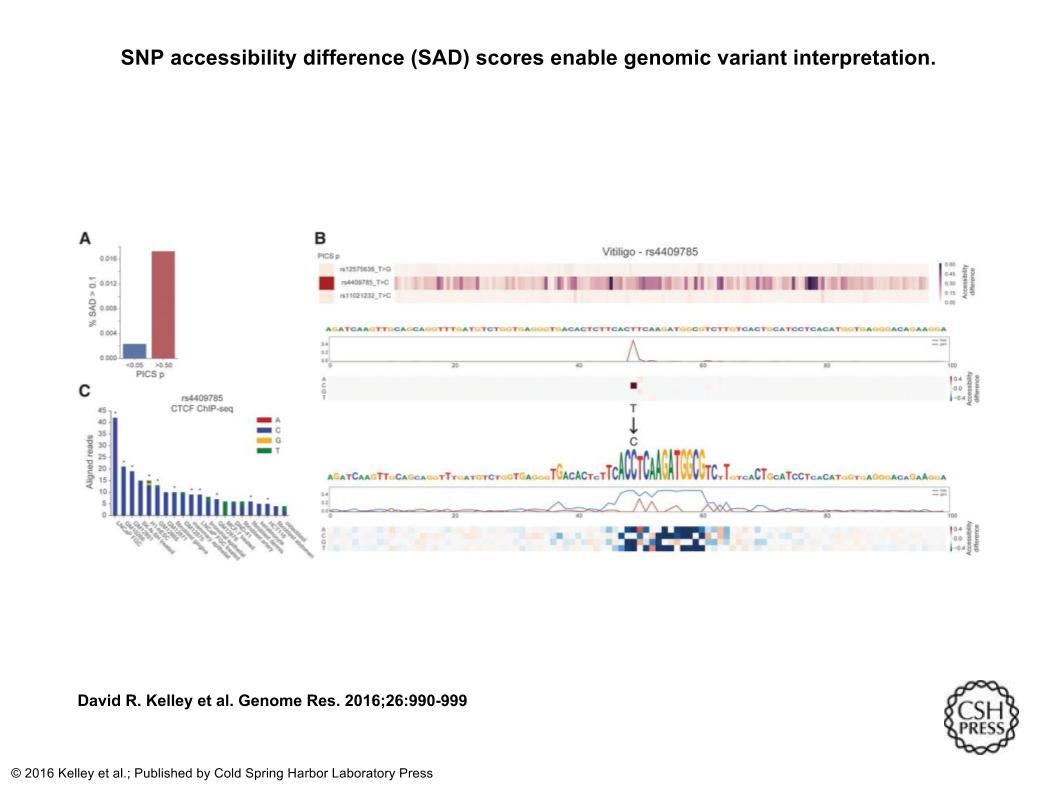
http://genome.cshlp.org/content/26/7/990.long
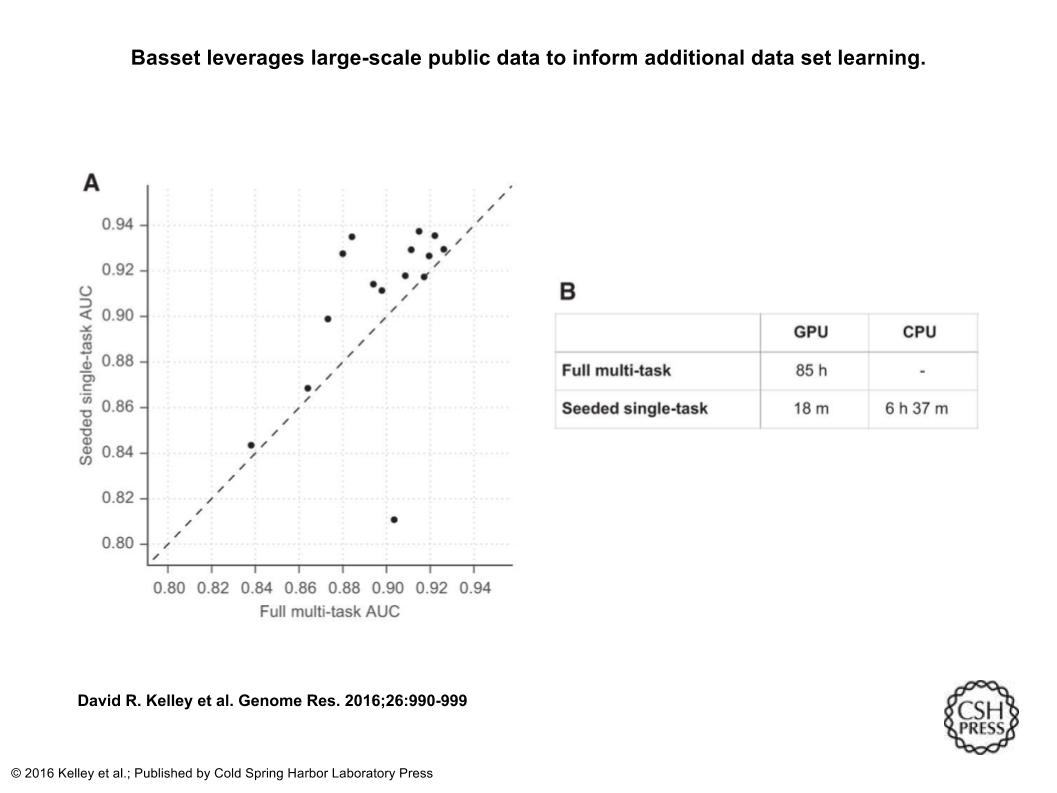
次のFig6について。(A)散布図は、x軸上の164個の細胞タイプすべてについて訓練されたフルモデルによって達成された15個のデータセットについてAUCを示し、y軸上にそのデータセットのみを調べる手順をシミュレートする手順によって得られたAUCを示す。データセットだけを調べるために、149個のセル(15個を除去した後)にモデルを事前訓練し、そのモデルのパラメータで追加セルをシードトレーニングし、新しいデータを1回トレーニングする。この迅速な処置は、他の同様の上皮細胞を用いたマルチタスク訓練が有益であった1つのデータセット(HRCEpiC、腎皮質上皮細胞)以外の全てにおいて有効でした。多くの細胞でAUCが改善されたことは、我々の完全なモデルが能力の増加または正則化の減少によって恩恵を受ける可能性があることを示唆している。(B)シードされたトレーニング手順は、GPU上ではるかに高速であり、実行可能なCPUトレーニングを可能にする。
http://genome.cshlp.org/content/26/7/990.long
理解するのも訳すのも大変でした。。まだまだわからないことだらけですが、今後もっとがんばりたいと思います。。
鈴木瑞人
東京大学大学院 新領域創生科学研究科 メディカル情報生命専攻 博士課程1年
東京大学機械学習勉強会 代表
NPO法人Bizjapan